 Share
Share




This article was co-authored with Jenny Yu, Chemical & Life Sciences Practice Leader for the UK and Ireland at Marsh. As this is a fast moving topic, please note that this article is current as at 18/09/2024. For further information, please contact Paula Margolis, Samantha Silver, or Jenny Yu.
This article follows Part 1: New frontiers in cell and gene therapies: A growing market
The unique and distinctive characteristics of cell and gene therapies (CGTs), also referred to as advanced therapy medicinal products (ATMPs) due to varying terminology from different regulators1, have created alternative regulatory approaches owing to the challenges of regulating them within traditional frameworks.
As discussed in our first article in this series, CGTs could potentially treat and cure diseases previously believed incurable. Therefore, the role of robust, effective regulation has become increasingly critical to advancing the safe development of CGTs, while also creating an environment conducive to innovation and commercial viability.
This article explores the challenges posed by the current regulatory frameworks governing CGTs in the UK, EU, and US, and how they are evolving to help foster innovation in the sector.
Regulatory frameworks: An overview
The broad and varied nature of CGTs has resulted in regulatory frameworks developing separately across different jurisdictions.
The UK
The UK has a comprehensive, stringent regulatory framework that aims to balance the need for effective governance and commercial development — partly comprising laws and regulations derived from EU law.
For CGTs, there are different regulators that work collaboratively to support establishments, including:
| The Human Fertilisation and Embryology Authority (HFEA) | Through the Human Fertilisation and Embryology Act (1990) and the Human Fertilisation and Embryology (Research Purposes) Regulations (2001), the HFEA regulates the use of human embryos or human admixed (human-animal) embryos to derive stem cells for use in the treatment of patients. |
| The Human Tissue Authority (HTA) | Through the Human Tissue Act (2004), the HTA regulates organisations that remove, store, and use human tissue or cells for a range of activities, including research and for use as starting materials for CGTs. |
| The Medicines and Healthcare Products Regulatory Agency (MHRA) |
The MHRA is responsible for clinical trials involving the therapeutic use of stem cells or stem cell lines, including the assessment of CGT applications for Clinical Trials Authorisation. Trials must be conducted in accordance with the Medicines for Human Use (Clinical Trials) Regulations (2004)2, if they present as exerting a pharmacological, immunological, or metabolic action. |
The implementation of new regulatory and licensing pathways has led to an acceleration in CGT approvals over the last decade. Since the UK’s approval of Holoclar in 2017, as of January 20243, 23 CGT products have been approved for use in the UK — with an additional 84 drugs also reportedly in clinical development4.
The EU
The European Medical Agency’s (EMA) Committee for Advanced Therapies is the multidisciplinary committee responsible for assessing the quality, safety, and efficacy of ATMPs.
ATMPs are regulated, like other pharmaceutical products, through a centralised marketing authorisation procedure, which provides a single evaluation and authorisation applicable to all EU countries. Companies developing ATMPs must comply with various regulations, requirements, and standards, including:
- The regulation on advanced therapy medicinal products5 (ATMP Regulation), which provides the overarching framework on ATMPs.
- Standards of good clinical practice when carrying out clinical trials in accordance with the EU Clinical Trials Regulation6 and other national requirements. There are also requirements on good manufacturing practice7.
- The EMA’s series of guidelines governing clinical trials of ATMPs.
The Tissues and Cells Directive8, which defines the safety and quality standards for tissues and cells intended for human applications. The Directive is due to be replaced by the proposed Regulation on substances of human origin which aims to strengthen the existing framework while providing more flexibility to account for scientific and technical developments.
Spotlight on HoloclarTo date, 24 ATMPs have received marketing authorisation in the EU, with Holoclar9 being the first to receive authorisation in 2014. This landmark development followed a long road to approval, with clinical testing beginning 17 years earlier in 1997. At that time, regenerative medicine was in its infancy and technologies still needed to be developed to make treatments more reliable. Notably, scientists involved in Holoclar’s development cited the introduction of the ATMP Regulation as hampering product development. While the regulation was necessary to ensure that such therapies were safe, ensuring compliance required the company to conduct further work and investigation. |
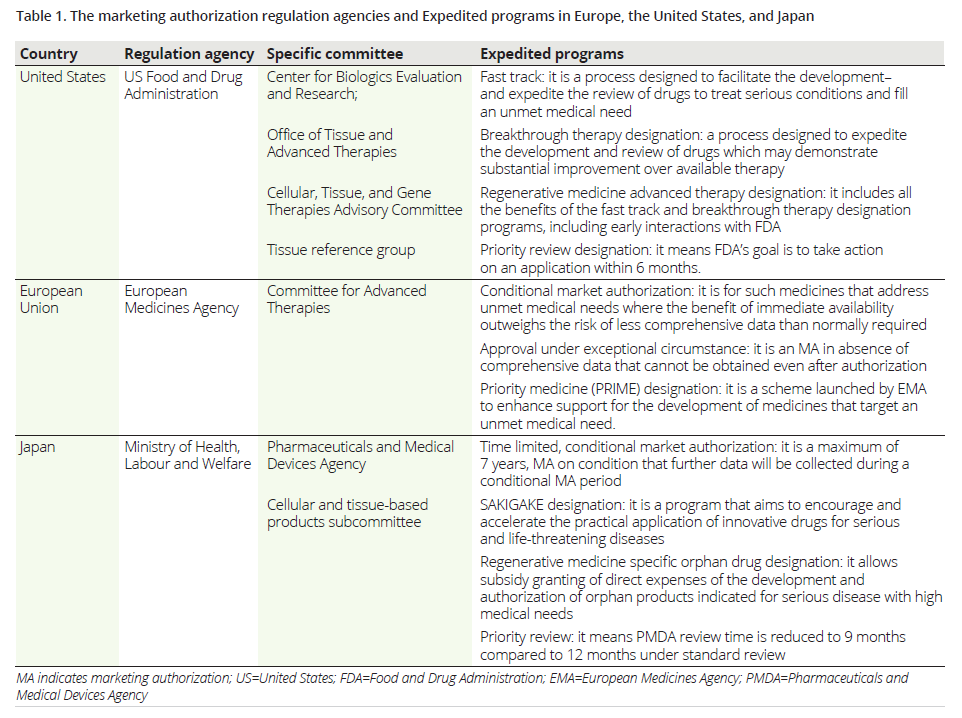
The US
The Food and Drug Administration’s (FDA) Center for Biologics Evaluation and Research regulates the development, testing, and review of CGTs.
CGTs are regulated as “biological products”10 and are, therefore, assessed according to the same regulatory standards and criteria for the quality, safety, and efficacy as biologics. CGTs require submission of an Investigational New Drug Application before initiating clinical studies and an approved Biologics License Application, in order to be marketed in the US.
Historically, CGT approvals by the FDA have taken a long time to come to fruition. It took the FDA 27 years to approve Luxturna, the first gene therapy11, following the initiation of the first approved study in 1990. This delay was due to various factors, including FDA requests for further studies and multi-centre trials.
To date, 37 CGTs have been approved by the FDA, of which seven were approved in 2023 alone. This trend is expected to continue on an upward trajectory as more clinical studies are initiated following the launch of FDA initiatives to expedite approvals. By 2025, he FDA expects to approve 10 to 20 CGTs a year.
Regulatory challenges
The differing and complex CGT regulatory frameworks pose significant challenges for manufacturers, including:
- Unsuitability of conventional regulatory requirements: CGTs are required to adhere to the same conventional requirements as other biopharmaceuticals — which are often unsuitable and can compromise the integrity of CGT modalities. Similarly, the introduction of novel starting materials, such as primary human cells, or the small-scale manufacturing process to meet one a single batch patient supply, come with challenges, such as those relating to product quality and viability, that cannot be properly addressed within current regulatory guidelines.
- Extensive post-approval studies: A comparative study of ATMP regulatory submissions with those for other biologics found that ATMP developers need to comply with more post-approval commitments to identify the long-term safety and efficacy of ATMPs. With many clinical and manufacturing objections for ATMPs reported to arise in the post-approval phase, this can impose a significant financial and administrative burden on ATMP developers which in turn, can hinder product performance and market access12.
- Adherence with evolving regulatory frameworks: Regulatory bodies have struggled to keep pace with advancements in CGT development, requiring them to adapt quickly to account for emerging scientific developments and ensure CGTs are developed in accordance with highest standards of safety and efficacy. This can be resource-intensive and costly.
- International differences in product classification and guidelines: Definitions of CGT products and their components differ between countries which can cause problems across the product lifecycle, particularly global clinical trials. Clear definitions are crucial to determining the application of the correct, corresponding regulation. Further, as countries continue to develop their own regulatory frameworks for CGTs, there is a risk of divergent guidelines developing across different regions, which in turn, is likely to pose significant challenges to sponsors with multinational development programmes.
Fostering innovation
Greater collaboration between industry stakeholders and regulators is enabling businesses to tackle regulatory challenges and foster innovation within the CGT sector. Efforts are increasingly being made to create regulatory pathways and initiatives that expedite the regulatory process and accelerate market approval of CGTs, examples include:
- The FDA’s Regenerative Medicine Advanced Therapy designation aims to expedite product development and submission review timelines — leading to patients receiving new medicines more quickly on a global scale. The FDA’s approval of Zolgensma in 2019, the first approved gene therapy to treat children under two years of age with spinal muscular atrophy, was subject to a series of designations designed to accelerate the regulatory process.
- The EU’s priority medicine designation provides opportunity for developers, in the early stages of product development, to have open communication with the EMA’s Committee for Medicinal Products for Human Use or the Committee on Advances therapies. This can optimise clinical development plans and accelerate the process for receiving conditional or full marketing authorisation approval, saving time and costs.
- Japan’s SAKIGAKE designation enables companies to access many development-supporting benefits, such as shorter lead times for regulatory consultation and quicker New Drug Application reviews. Japan has also aligned its regulations with foreign regulators, including the FDA and EMA, to ease the regulatory pathway for international developers.
- In the UK, the NHS Accelerated Access Collaborative and the MHRA’s Innovative Licencing and Access Pathway (ILAP) reduce the time to market access for innovative medicines. The UK became the first country in the world to approve Casgevy13 in 2023, via an Innovation Passport granted under the ILAP.
Looking ahead, continued collaboration between relevant stakeholders, such as manufacturing companies and regulators, is key to establishing workable regulatory frameworks and best practices as early as possible. This will help drive the commercial success of these innovative and life-changing therapies.
This article follow Part 1: New frontiers in cell and gene therapies: A growing market
In the next article of our series, we consider the impact of CGTs on the insurance market and the potential legal risks.
1 The EMA uses the term ATMP instead of CGT. ATMPs are further categorised into gene therapy medicinal product (GTMP), somatic cell therapy medicinal product (SCTMP), tissue-engineered therapies (TET), and combined advanced therapies.
2 The MHRA has proposed to update the regulatory framework governing clinical trials to make it faster and easier to gain approval and run clinical trials in the UK.
3 BIA Report: UK cell and gene therapy: Leading the path to transformative medicine; published January 2024.
4 Data as of 31 October 2023.
5 Regulation (EC) No 1394/2007 which amended the Directive on the Community code relating to medicinal products for human use (2001/83/EC) and the Regulation laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency Regulation (EC) No 726/2004, with regards to ATMPs.
6 Regulation (EU) No. 536/2014.
7 Commission Directives 2003/94/EC and 2005/28/EC.
8 Directive 2004/23/EC.
9 Defined in section 351(i) of the Public Health Service Act (PHS), 42 U.S.C. 262 (i).
10 Voretigene neparvovec-rzyl developed by Spark Therapeutics.
11 An advanced medicinal product based on autologous stem cells and capable of restoring sight in burns victims.
12 Elsallab M., Bravery C.A., Kurtz A., Abou-El-Enein M. Mitigating deficiencies in evidence during regulatory assessments of advanced therapies: A comparative study with other biologicals. Mol. Ther. Methods Clin. Dev. 2020;18:269–279.
13 Exagamglogene autotemcel (exa-cel) is a non-viral cell therapy sold under the brand name, Casgevy.
 Life sciences
Life sciences
 Insurance and reinsurance
Insurance and reinsurance
{kind=link}